Part III
Chapter 15
Findings of the RPRP
Chapter 15: Findings of the RPRP
In this section, we present the results of the RPRP. We begin with a general characterization of our overall assessment of the research documents. We also provide additional analysis of the impact of the level of risk and kind of experiment (nonradiation vs. radiation) on our evaluations. Next, we turn to a discussion of what the Committee found most troubling in these documents, organized around issues of understanding, voluntariness, and decisional capacity. Finally, we look at problems that were common in the sample as a whole, including the readability of consent forms and deficiencies in documentation.Overall Assessment
Reviewer teams registered their overall assessment of each set of documents using a scale from 1 to 5, where 1 was taken to indicate no ethical concerns and 5 was taken to indicate serious ethical concerns. This scoring scale was used to assist reviewers in organizing their overall evaluations of the set of documents for each research proposal. These ratings were made in concert by the two reviewers after each had completed his or her own independent review. Ratings of 4 and 5 are grouped together in the discussion that follows because reviewers generally did not differentiate between the two; both ratings were used when documents raised serious ethical concerns for reviewers.[19]
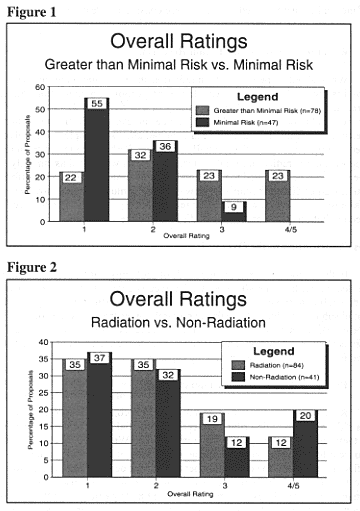
For the total sample of documents from 125 radiation and nonradiation research proposals, two-thirds received ratings of either 1 (34%) or 2 (34%), while 18 percent received a rating of 3 and 14 percent received a rating of 4 or 5.
Level of Risk
Reviewers identified whether the research proposals as described in the documents involved minimal risk or greater than minimal risk of harm to subjects; 78 proposals were considered to involve greater than minimal risk (including 24 proposals that were evaluated as "maybe" greater than minimal risk[20]risk" did not significantly affect the proportion of studies that received each overall rating, the Advisory Committee decided to evaluate these two groups of studies together.), while 47 proposals were considered to involve minimal risk.
There was a marked difference in the distribution of ratings between minimal-risk and greater-than-minimal-risk studies (Figure 1). Although a substantial number of greater-than-minimal-risk studies received ratings of 1 or 2, all of the studies that received 4s and 5s were considered greater than minimal risk.
Radiation versus Nonradiation Research
While about 70 percent of both radiation and nonradiation proposals received ratings of 1 or 2, a somewhat higher proportion of nonradiation studies than radiation studies received overall ratings of 4 or 5 (Figure 2). This difference could not be explained by differences in level of risk; the proportion of studies in the nonradiation subsample and the radiation subsample that involved greater than minimal risk was essentially the same. Perhaps the lower proportion of proposals in the radiation sample whose documents were rated as ethically problematic can be attributed to the second layer of scrutiny that is often afforded radiation studies during the initial review process. It must be noted, however, that because there were few studies that received ratings of 4 or 5, differences between radiation and nonradiation studies may not be significant.
Issues Contributing to the Overall Ratings
In this section we examine the kinds of problems that troubled reviewers in the documents from the 40 proposals that received ratings of 3, 4, or 5. These problems fell in to three categories: (1) factors likely to affect the adequacy of potential subjects' understanding of the research (other than questions of competence); (2) factors likely to affect the voluntariness of potential subjects' decisions about participation; and (3) approaches to the inclusion of subjects with limited or questionable decision-making capacity.
Factors Likely to Affect Understanding
Reviewers were likely to give a 3, 4, or 5 to proposals whose consent forms did a poor job of describing either what potential subjects stand to gain or what they stand to lose by participating in research. We looked carefully at how the consent forms presented the purpose of the study, its potential for direct benefits to the subject, the distinction between direct benefits and benefits to medical science, and alternatives to participation. How well consent forms communicated the realities of what it would be like to participate in the proposed research, including the likely impact on quality of life, also came under scrutiny. We were troubled, for example, by consent forms that, when compared with the information provided in the grant proposal or other research documents, appeared to overstate the therapeutic potential of research, either explicitly or indirectly.
This issue was of particular concern to the Committee when the subjects being recruited were patients with poor prognoses. For example, one study, which was presented as primarily a toxicity study in the accompanying research documents, was cast differently in the consent form: "One objective is to find out how well patients respond to treatment. . . . If treatment works in your case, it may shrink your tumor or cause it to temporarily disappear, and/or prolong your life and/or improve the quality of your life. . . . Another objective of this study is to find out what kind of side effects this treatment causes and how often they occur."[21]
There also was significant concern about the use of the word treatment in consent forms for pharmacological studies. Phase I studies are designed to establish the maximum tolerated dose (MTD) for new chemotherapeutic agents and radiation regimens, which are then subjected to limited (Phase II) and then more extensive (Phase III) clinical trials to determine therapeutic effectiveness.[22] Although some Phase I studies contain elements of Phase II research and can appropriately be characterized as holding out at least a remote prospect of benefit to the subject, for some Phase I studies even the suggestion that subjects might benefit is inappropriate.
Reviewers were influenced in their overall assessments by inadequate descriptions of the physical risks of participating in the research. Reviewers were concerned, for example, when consent forms did not discuss the risks potential subjects faced in being removed from their standard treatments to be placed on an experimental protocol. In one study, patient-subjects were taken off cardiac medication in order to participate in a diagnostic study that offered no direct benefit to them. Any risks involved in the removal from this cardiac medication were not addressed in the consent form. The Advisory Committee also identified consent forms in which the possible lethality of drug treatments and radiation exposures was not adequately discussed. This occurred in contexts where patient-subjects generally faced far greater risks from their underlying illnesses, but, nevertheless, we felt that the consent forms should have been more forthcoming. A number of projects that involved combination drug treatments, for example, did not provide the potential subject with an estimate of the possibilities of death or major toxicities from a combination of drugs. One study involved a combination chemotherapy consisting of twelve different drugs but did not address the uncertainty of risk resulting from this new and investigational combination. Although the hazards and side effects for each drug were described individually, there was no discussion of overall risks and harms.
Even where consent forms described the risks of the research, there was often little mention of how participation would affect the subject's ability to function in daily life or how ill subjects might be made to feel during the course of the research. This omission was of particular concern to us when the implications for quality of life were markedly different depending upon whether a person decided to participate in the research or accept standard medical management, such as when standard management included only palliative care or watchful waiting. In one end-stage cancer study, for example, the consent form stated only that there may be "[o]ther general complications which may occur from combinations of chemotherapy drugs, including weight loss and loss of energy." The Advisory Committee was troubled that in such studies patient-subjects may not understand that although the research protocol might offer a chance to extend life, the time gained might be compromised by additional limitations in the quality of life resulting from participation in the study.
Reviewers also noted a number of problems in some consent forms for randomized clinical trials. For example, when some patient-subjects were randomized to receive the standard treatment while others would undergo an experimental procedure, reviewers commented that physical risks associated with the standard treatment or procedure were sometimes not adequately addressed in the consent forms. In one study of the effectiveness of a new compound for the decontamination of people who had ingested a radioisotope, although the grant proposal indicated that subjects would be randomized to receive a placebo, this information was not included in the consent form. In fact, the consent form only vaguely discussed the experimental procedures. "I [subject name] authorize [physician name]. . . to administer decorporation therapy utilizing the drug [name of drug]."
The Committee recognizes the difficulties facing investigators in communicating to potential research subjects a complex set of experimental procedures, side effects, long-term risks, trade-offs relative to alternatives, and other relevant information. This task is not impossible, however. We reviewed documents from several complex research proposals that at the same time had excellent consent forms.
For example, we reviewed documents from a proposal for a Phase I study of experimental antibody therapy that involved a number of possible risks; imposed a number of inconveniences including restrictions on sexual activities and a weeklong time commitment; and, as a Phase I study, offered little prospect of direct benefit to subjects. The consent form for this study addressed each of these issues in understandable language, briefly described how the monoclonal antibodies used in this research were derived, and explained that the U.S. Food and Drug Administration (FDA) permits experimental, new forms of therapy to be tested in a limited number of patient-subjects in Phase I studies. This consent form presented enough useful information to enable potential subjects to make an informed decision about whether to participate in the research, and it was not overly optimistic about the prospect of direct benefit to the patient-subject.
Another complex, greater-than-minimal-risk study with a good consent form involved an investigational radiation treatment, radiosurgery, for patient-subjects who had vascular disorders of the brain. The consent form for this study described the experimental procedures step-by-step with a very realistic picture of what participation would entail. Potential risks, possible benefits, and alternatives to participation for this experimental therapy were clearly presented. Furthermore, information that was likely to discourage some patients from enrolling--the possibility that participation in this study might limit the effectiveness of similar types of radiotherapy for the patient in the future--was disclosed in the consent form.
Factors Likely to Affect Voluntariness
As is discussed later in this chapter, the documents we reviewed often provided no basis on which to judge whether the participation of potential subjects was likely to be voluntary or not. In some cases, however, the information provided was sufficient to raise concerns. One was a neuroscience study that offered no prospect of medical benefit to potential subjects. Subjects were being recruited from among former cocaine addicts who were living in a residential treatment facility. Although compensation was not needed to reimburse subjects for travel expenses or loss of income, subjects were being offered $100 to participate. Reviewers were concerned that this cash payment might make it easier for those people struggling to break an addiction to get cocaine. Moreover, as part of the study, cocaine was injected into the body in order to measure brain uptake. Even if this procedure was not likely to have a physiologic effect upon the subjects, we were concerned that subjects may have been encouraged to participate because the research involved the injection of cocaine. We were also concerned about how their receiving cocaine as part of the research might affect the subjects' perceptions of themselves during the recovery process.
By contrast, the following text from a consent form for employee-subjects (colleagues of the investigators) who are smokers illustrates exemplary handling of the voluntariness issue in a minimal-risk study. The study, which involved no risk of physical harm to the subjects, was designed to measure environmental tobacco smoke.
Your participation in the experiments is entirely voluntary and you are free to refuse to take part. You may also stop taking part at any time. Because you are a colleague here at [research institution], we want to be especially clear on this point. We have approached you about the possibility of your volunteering for these experiments. Your refusal to participate or to continue will not be questioned by us, nor will it (or should it) be discussed further with anyone else.
Inclusion of Subjects With Limited Decisional Capacity
Several issues revolve around how certain factors that influence a subject's decisional capacity may affect his or her ability to understand the implications of participating in research. There is, for example, considerable controversy over how to conduct research ethically in emergency medicine when, because of the acute nature of the medical problem, the patient is temporarily incapacitated and no family members are available for consultation. The documents of one proposal raised some of these issues. In this example, 5 minutes were allotted to obtain consent from subjects who were recruited in the emergency room while their chest x-ray films were being processed. Under the stressful conditions of an emergency room and while experiencing chest pain, the decisional capacity of potential subjects was likely to be severely compromised. Reviewers expressed concern about the subjects' ability, in such a context, to comprehend the study adequately and then make a voluntary decision about whether or not to participate. In another study, women in preterm labor were recruited to participate in a study that involved collecting data about the infants born to these women. Although the proposal stipulates that "[n]o mother will be approached while under undo[sic] stress or in excessive pain," reviewers were nonetheless concerned about consent having been solicited during preterm labor.
The Advisory Committee also reviewed the documents of studies involving children and adults with questionable decision-making capacity, several of which raised serious ethical concerns.
Sixteen of the studies included in the Advisory Committee's review involved children as research subjects; 11 of these 16 studies, according to federal regulations, should have had assent forms as well as parental permission forms.[23] The documents we received on each of these proposals all included parental permission forms. We received assent forms for 8 of these 11 proposals. The 3 studies for which we did not receive assent forms all involved greater than minimal risk, 1 of which may not have offered any prospect of medical benefit to the children-subjects.
This last study illustrates a major issue in the ethics of research involving children. Current regulations permit the use of children as subjects in research that offers no prospect of direct medical benefit to them when the research poses no more than minimal risk. Nontherapeutic research on children posing more than minimal risk is permitted under special circumstances. A central, unresolved question is whether the administration of tracer amounts of radioactive materials to children can properly be classified as a minimal-risk intervention.
Eight studies in the project sample sought to recruit adult subjects with questionable decision-making capacity. 6 of the 8 appeared not to offer potential medical benefits to the subjects; two of the 6 were epidemiologic studies.
The Committee's concerns focused primarily on the remaining four studies, all of which involved diagnostic imaging with cognitively impaired persons, such as those with Alzheimer's disease. The imaging processes required that the subjects' movements be restricted, yet there was no discussion in the documents or consent form of the implications for the subjects of these potentially anxiety-provoking conditions. Nor was there discussion of the subjects' capacity to consent or evidence that appropriate surrogate decision makers had given permission for their participation. We were particularly troubled that two of these studies exposed subjects to greater than minimal risks. The question of whether or under what conditions adults with questionable decision-making capacity can be used as subjects of research that offers no prospect of benefit to them is unresolved in both research ethics and regulation. When such research puts potentially incompetent people at greater than minimal risk of harm, it is even more ethically problematic.
Common Problems With the Documents
We turn now to a discussion of issues that emerged often in the documents we reviewed, including documents that raised only minor concerns.
Consent Form Language
Although inappropriate reading level in a consent form was generally not sufficient in and of itself to result in ratings of 3, 4, or 5, it was sufficient for a rating of 2. A significant majority (nearly 80%) of the proposals receiving a 1 included consent forms that used a reading level appropriate for the study population. By contrast, the reading level was judged to be appropriate in no more than half of the remaining consent forms.
Reviewers raised a number of issues that they felt may have contributed to problematic reading levels in the consent forms. One such issue pertains to the complexity of the research being proposed. We were disturbed to find that in their attempts to convey complexities to the subject, investigators often drafted consent forms that were too lengthy, highly technical, and generally unintelligible. Consider the following, for example: "The purpose of this study is to obtain a 'map' of brain cholinergic receptors. . . . This is done by administering, intravenously, small amounts of a radioactive substance that attaches to brain acetylcholine receptors and then producing a map of these receptors using Single Photon Emission Computed Tomography (SPECT)."
Still another consent form included language such as "[y]ou will then be positioned in a recumbent position," and "[a]nother possibility is poor regional function because of ongoing or intermittent ischemia at rest, resulting in anginal symptoms and global function that is worse than it can or should be."
A number of the consent forms included standard ("boilerplate") language that was often in a smaller type and distinct from the rest of the document. The presentation of information in this manner may have given subjects the impression that the information was less important and easily skipped. Sometimes these sections contained the only discussion of such critical topics as alternatives to participation, costs to the subject, confidentiality, potential benefits of participation, and voluntariness of participation.
The Advisory Committee found that intramural institutions often used a standard consent form that contained boilerplate language provided by their respective agencies. The following passage is an example of such language. It appeared in smaller type at the top of consent documents, clearly separated from the rest of the text:
We invite you (or your child) to take part in a research study at the [named institution]. It is important that you read and understand several general principles that apply to all who take part in our studies: (a) taking part in the study is entirely voluntary; (b) personal benefit may not result from taking part in the study, but knowledge may be gained that will benefit others; (c) you may withdraw from the study at any time without penalty or loss of any benefits to which you are otherwise entitled. The nature of the study, the risks, inconveniences, discomforts, and other pertinent information about the study are discussed below. You are urged to discuss any questions you have about this study with the staff members who explain it to you.
Reliance on Disclosures Not Subject to IRB Review
When patients are being approached to participate in research that has implications for the medical management of their illness, it is understandable and indeed desirable that patient-subjects discuss the proposed research with their treating physician. The Committee was disturbed, however, when consent forms indicated that the only presentation to potential subjects of key information about the research was to take place in such undocumented discussions. This suggests that it is difficult, if not impossible, for IRBs to judge whether potential subjects were being provided an adequate base of information on which to make an informed decision. There is no documentary record, either in the consent form or in other materials submitted to the IRB, of what potential subjects have or will be told about key aspects of research participation.
In some cases, consent forms indicated that subjects themselves were responsible for approaching physician-investigators for explanations of the choices available and guidance on how to compare the experimental protocol to standard treatment. Consider the following example:
Your (child's) doctor can provide detailed information about your (child's) disease and the benefits and risks of the various options available. You are (your child is) encouraged to discuss this with your (child's) doctor.
In this instance, it is unclear whether the phrase your doctor refers to the patient-subject's personal physician, a physician who is a member of the research team, or a physician who is both. This passage, as well as passages in several other consent forms, suggests that conversations between subjects and the doctors occurred after consent was given. Examples of this follow: "Severe and sometimes deadly side effects have occured when high doses of this drug have been given . . . You and your doctor will determine whether the benefits of such treatment outweigh the risk"; and "You will discuss the options with your physician and decide between . . . [surgical alternative] . . . or [medical alternative] . . ." Subjects in these studies may have received information critical to their decision making process only after giving their consent to participate in the research and without the IRB knowing the content of that information. This is particularly troublesome because these statements comprise the only discussions of side effects and alternatives, respectively, in these consent forms.
Other consent forms seemed to rely on disclosures that had already taken place by the time potential subjects were approached to give their consent, and so could not be afforded IRB review. One such consent form began, "The following is a summary of the information your doctors gave you when discussing this treatment with you. Please read it and ask any questions you may have." The summary that followed provided little specific detail. The Committee was left wondering whether the IRB was in a position to make a judgment about the adequacy of this prior disclosure.
Voluntariness
If an informed consent is to be a meaningful act of decisional autonomy, it is essential not only that the consent be based on adequate understanding but also that it be substantially free from coercive or manipulative influences. We found, however, that many proposal documents, including applications to IRBs, did not contain enough information to make a judgment about the likely voluntariness of subjects' consent decisions. For example, there was often insufficient or no information about who was soliciting a potential subject's consent and under what conditions.[24]
Often the only information in the documents reviewed that bore on issues of voluntariness was the inclusion in consent forms of boilerplate language to the effect that participation was voluntary. In most cases, the issue of voluntariness was simply ignored in proposal documents submitted to the IRBs and funding agencies, precluding us (and, presumably, IRBs) from making any judgments about the procedures employed to ensure voluntary decision making.
Scientific Merit
A controversy has long existed over whether the role of IRBs includes evaluation of the scientific merit of proposed research. Some argue that evaluation of scientific merit lies outside the scope of IRB review, while their opponents contend that it is impossible to do a proper assessment of the benefit-risk ratio without evaluating the potential contribution to science. Based on the documents we received, it was sometimes difficult to make judgments about scientific merit. In some cases, reviewers felt that they could not establish from the documents available to them whether there was sufficient scientific merit to warrant the exposure of human subjects to risk or inconvenience.
Psychosocial and Financial Risks
In research where psychosocial risks were clearly an issue, these risks were often inadequately addressed in proposal documents. A number of proposals that included neuropsychological batteries, for example, failed to discuss the potential anxieties that may result from participation in the study. The objective of one research project involved the inducement of sadness in the subject. Neither the consent form nor the research documents addressed the possibility that the sadness would not resolve itself quickly and that psychological counseling or other therapy might be necessary.
Four studies reviewed by the Advisory Committee involved DNA screening to determine the subjects' carrier status for a particular gene. None of the proposals for these studies addressed the potential psychosocial impact of learning about one's carrier status, including possible implications for other members of the subject's family or the potential for insurance discrimination. The availability of genetic counseling for these subjects was not mentioned in consent forms. Reviewers also were concerned that some proposals did not clearly explicate the types of tests that were included in what was referred to as "chronic disease screening" in the consent forms. This lack of specificity was particularly troubling for "chronic disease screens" that included human immunodeficiency virus (HIV) testing. Although the anxieties and social risks of HIV testing were likely to be addressed on a separate HIV-specific consent form, any study that requires HIV screening as part of its eligibility criteria should make that clear to subjects so that those who do not wish to undergo HIV testing can decline participation.
Another area that was sometimes inadequately described in both consent forms and research documents was the financial cost to the subject of participating in the research. Costs were often briefly addressed in the boilerplate section of the consent form, but usually no project-specific information about actual expenses was offered to the subject. Reviewers were concerned that subjects might not appreciate the real costs and the possibility that insurance companies would be very reluctant to cover them. This omission was particularly troubling in studies involving seriously ill patient-subjects who may be at risk of spending much of their assets on research interventions at the end of life.
Justice in the Selection of Subjects
Most research documents did not include specific information about the subject populations that would be involved in the protocol. Unless IRBs are receiving more information on this topic than that provided in the documents reviewed by the Advisory Committee, they are clearly ill-equipped to address the social policy goal[25] of including women, minorities, and other groups in research. The racial and ethnic composition of the subject sample, for example, was specified in only one-quarter of the proposals whose documents were reviewed by the Committee.
The only frequently mentioned reason for excluding a person from participation in research was pregnancy. Pregnant women were explicitly excluded in 58 percent of the studies (73 of 125) and were explicitly included in only 5 percent (6 of 125) of the proposals. Pregnancy tests were often included in the eligibility screening procedures for women who were willing to participate in research. The RPRP sample also included 13 studies in which women who were not pregnant were expressly excluded from participation. There was no scientific reason to exclude women as subjects of research in any of these proposals. In two of these instances, women were excluded expressly because of the possibility that they might become pregnant.
The Committee's interpretation of the implications of these findings can be found in the "Discussion" section at the end of the chapter.


